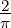 arctan
arctan 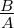 is analogous to BAF but is not truncated (unlike BAF,
which is typically truncated to the interval [0,1], with homozygotes often having BAF values
of 0 or 1).
is analogous to BAF but is not truncated (unlike BAF,
which is typically truncated to the interval [0,1], with homozygotes often having BAF values
of 0 or 1).
The HI-CNV software calls copy-number variants (CNVs) from SNP-array genotyping probe intensity data in large cohorts. HI-CNV uses a haplotype-informed approach that improves power to detect shorter CNVs (spanning as few as two genotyping probes) by incorporating probe data from multiple individuals who share an extended SNP haplotype—and are thus likely to also share any CNVs co-inherited within these shared genomic segments.
The HI-CNV algorithm is described in:
Hujoel MLA, Sherman MA, Barton AR, Mukamel RE, Sankaran VG, Terao C, Loh PR. Influences of rare copy number variation on human complex traits. Cell (2022).
You can download the latest version of the HI-CNV software at:
Previous versions are available in the old/ subdirectory.
On most Linux systems, installation should only require unpacking the HI-CNV_vX.X.tar.gz download package. The bin/ subdirectory of the package contains precompiled standalone (i.e., statically linked) 64-bit Linux executables for all steps of the HI-CNV pipeline that should work on most systems.
If you wish to compile components of the HI-CNV software package from the source code (in the src/ subdirectory), you can do so using the compile.sh script provided in the src/ subdirectory. You will need to edit the compile.sh script to specify local paths to the following libraries:
For reference, the provided executables were created on the Harvard Medical School “Orchestra 2” research computing cluster using GCC 4.8.5 and were linked against the Boost 1.58.0 library and Intel MKL 2019 Update 4.
HI-CNV requires the following input data:
Sample information is provided in a single file, while all other data are split by chromosome (i.e., 22 files for chr1-22). File formats for all inputs are detailed below.
The sample info file (plain text, whitespace-delimited) should begin with the following 5-column header line:
ID_1 ID_2 ID_imp genotypingSet inUniformAncestrySet
Subsequent lines should provide the following information about each individual contained in the SNP-array genotyping data files:
SNP-array genotype calls (e.g., generated by Illumina or Affymetrix) should be provided in a single PLINK file set per autosome (i.e., 22 .bed/.bim/.fam files in total). If different samples were genotyped on different SNP-array platforms, genotype call files should be merged using plink, and the genotyping sets should be indicated in the genotypingSet column of the sample info file (see section 3.1).
Genotyping probe intensity data should be provided in two files per autosome (2 x 22 files total) containing measurements of:
arctan is analogous to BAF but is not truncated (unlike BAF,
which is typically truncated to the interval [0,1], with homozygotes often having BAF values
of 0 or 1).For each autosome c, the LRR and the θ file should each be provided in the following whitespace-delimited text format (.txt.gz):
HI-CNV requires phased SNP haplotypes in order to incorporate genotyping probe intensity data from multiple individuals who share an extended SNP haplotype (likely to have been co-inherited from a recent common ancestor). Such SNP haplotypes can be estimated by performing statistical phasing on the SNP-array genotype calls (e.g., using Eagle, SHAPEIT, or Beagle).
Phased haplotypes should be provided to HI-CNV in HAPS/SAMPLE format (one pair of files per autosome); conversion from other formats can be performed using bcftools or plink2.
HI-CNV also requires genotype information for variants near the sites targeted by SNP-array probes: such variants can impair probe hybridization (producing intensity signals similar to deletions), such that it is beneficial to mask probe data potentially affected by nearby variants.
Genotypes of variants not directly genotyped on a SNP-array are commonly estimated via statistical imputation using sequenced reference panels. Such imputed data is sufficiently accurate for the purpose of masking probe measurements in HI-CNV, so we recommend using previously-generated imputed data (e.g., from GWAS pipelines) if available.
HI-CNV requires imputed data to be provided in BGEN/SAMPLE v1.2-format, so if imputed data is available in VCF format, then conversion to BGEN v1.2 will need to be performed. However, because data from relatively few imputed variants are needed (only those very close to array SNPs), this conversion should be straightforward and space-efficient and can be accomplished using the following code:
### create UCSC BED file listing regions near SNPs on array
awk -v oligoLen=50 -v OFS=$’' t’ ’{
start=$4-oligoLen; end=$4+oligoLen-1;
if (start<=lastEnd) start=lastEnd;
print $1,start,end;
lastEnd=end;
}’ '
$INPUT_DIR/chr$CHR.bim '
> $INPUT_DIR/chr$CHR.imp.bed
### extract imputed variants in regions
bcftools view $VCF '
-R $INPUT_DIR/chr$CHR.imp.bed '
-Ob -o $INPUT_DIR/chr$CHR.imp.bcf
### convert to BGEN v1.2 8-bit format
plink2 --bcf $INPUT_DIR/chr$CHR.imp.bcf dosage=HDS '
--export bgen-1.2 bits=8 --out $INPUT_DIR/chr$CHR.imp
Additional notes:
The HI-CNV software pipeline requires running a series of steps described in detail below. To make running these steps as simple as possible, we have provided a set of scripts in the top-level directory of the download package, each of which runs one step of the pipeline.
The easiest way to run the scripts is to first create an analysis directory (anywhere on your system) with input/ and output/ subdirectories. Then, in the input/ subdirectory, create the following symbolic links to the input data described in section 3:
The HI-CNV scripts can then be run with the analysis directory provided as a single argument, e.g.:
./1_check_inputs.sh /path/to/analysis/dir
If no directory path is provided, then analysis will be performed in the HI-CNV download directory (assuming that inputs have been supplied in the input/ subdirectory; this subdirectory only contains placeholder symbolic links when initially unpacked).
Most steps of the pipeline (except 2_denoise_lrr.sh) analyze each autosome independently, so if you are running analyses on a compute cluster or cloud, you may wish to modify the scripts to submit parallel jobs, one per autosome. The final analysis step (9_call_CNVs.sh) can be further parallelized across different values of the IBD parameter.
This script invokes the check_inputs executable to run a series of checks on all input files verifying that they are properly formatted and consistent with one another. The check_inputs executable exits with an ERROR message if it detects an input problem that prevents analysis from proceeding. It reports WARNING messages if the input data has properties that are unexpected but do not necessarily preclude analysis. These WARNING messages should be carefully considered before proceeding, as they may indicate issues with data preparation or QC that need to be resolved first.
In addition to printing diagnostic output, the check_inputs executable also generates the following files that may be helpful for debugging:
This step of the pipeline is computationally light: a few hours per chromosome for a large biobank (N = 500,000 samples), with minimal memory use because input data are streamed.
The 1_check_inputs.sh script directs all of the above output files to be written to the output/check_inputs/ subdirectory of the analysis directory.
This script invokes the denoise_lrr executable to remove technical noise in total genotyping intensity (LRR) measurements captured by the top P principal components of the LRR matrix. The number of PCs is specified as a command-line argument; in our experience, P =50 is often a reasonable choice.
LRR PCs are computed once per genotyping set on up to 5,000 individuals randomly selected from among individuals belonging to the inUniformAncestrySet (see section 3.1). Denoised LRR is then computed for all individuals in the genotyping set by subtracting components of LRR along the top P PCs. To reduce risk of PCs capturing LRR variation arising from real copy-number variation rather than technical variation, PCs are computed using SNP-array data from all autosomes at once. Consequently, this step of the pipeline runs on a single compute node and has more substantial resource requirements:
The 2_denoise_lrr.sh script directs temporary and denoised LRR output files to be written to the output/array_data/ subdirectory of the analysis directory. A file containing estimates of relative LRR noise per sample (used to create sample batches in future steps) is also written to this directory.
This script invokes the merge_lrr_theta_geno executable to merge three types of SNP-array data for downstream analysis: (i) denoised LRR computed in the previous step, (ii) θ (discretized into 1-byte values), and (iii) genotype calls.
This step is computationally light: on the order of 1 hour per chromosome for a large biobank, with minimal memory use because input data are streamed.
The 3_merge_lrr_theta_geno.sh script directs the merged SNP-array data files to be written to the output/array_data/ subdirectory of the analysis directory.
This script invokes the call_CNVs_prelim executable to identify an initial reference set of large CNVs based on only total genotyping probe intensity (LRR) signal.
This step is computationally light: <1 hour per chromosome for a large biobank, with modest memory use (typically <8 GB) because each autosome is analyzed independently and input data are processed in batches. The large CNV calls generated in this step are used only to estimate genotype cluster parameters for CNV genotypes in the next step of the pipeline.
LRR-based CNV calling requires estimates of the mean LRR within deletions and duplications, which vary across SNP-array data sets and may not be known a priori. Consequently, the LRR-based CNV-calling algorithm takes as input initial guesses of mean LRR within DELs and DUPs but then refines these estimates using an iterative expectation-maximization (EM)-like approach (such that the overall analysis is robust to error in initial guesses). The number of iterations to run is specified via a command-line argument; typically a single iteration suffices.
The 4_call_CNVs_prelim.sh script directs the preliminary LRR-based CNV calls to be written to the output/prelim_CNV_calls/ subdirectory of the analysis directory.
This script invokes the compute_ref_clusters executable to estimate parameters of DEL and DUP genotype clusters (A, B; AAA, AAB, ABB, BBB) for a subset of SNPs with sufficient reference data available from large CNV calls from the previous step. Clusters are modeled as bivariate normal distributions in (θ, LRR) space, and bivariate normal parameters are estimated from observed genotyping probe intensities within large CNV calls. To account for technical variation in genotyping array measurements, samples are subdivided into batches based on genotyping set and LRR noise, and cluster parameters are estimated independently for each sample batch.
This step is computationally light: on the order of 1 hour per chromosome for a large biobank, with minimal memory use because input data are streamed.
The 5_compute_ref_clusters.sh script directs the reference genotype cluster data to be written to the output/snp_clusters/ subdirectory of the analysis directory.
This script invokes the predict_clusters executable to predict genotype cluster parameters at all SNP-array probes (leveraging genotype cluster patterns observed at reference SNPs for which CNV genotype clusters could be estimated in the previous step).
This step is computationally light: on the order of 1 hour per chromosome for a large biobank (assuming multithreading is used), with modest memory use (on the order of 1 GB for a SNP-array with 1 million probes).
The 6_predict_clusters.sh script directs the predicted genotype cluster data to be written to the output/snp_clusters/ subdirectory of the analysis directory.
This script invokes the find_IBD executable to compute each sample’s “haplotype neighbors”: i.e., for each haplotype of each individual, at each genomic position, which other haplotypes share the longest identical-by-descent (IBD) matches spanning the position. Haplotype matches are identified using a seed-and-extend approach that begins by using the positional Burrows-Wheeler transform (PBWT) to rapidly identify long exactly-matching haplotype segments; matches are then extended to account for genotyping errors and gene conversion events that can interrupt IBD segments.
This step is computationally moderate: on the order of 1 hour per chromosome for a large biobank (assuming multithreading is used), with moderate memory use (10-50 GB for a large biobank).
The 7_find_IBD.sh script directs haplotype match data to be written to the output/IBD/ subdirectory of the analysis directory.
This script invokes the make_imp_masks executable to flag SNP-array probe intensity measurements that may be compromised by the presence of nearby inherited variants that interfere with oligonucleotide probe binding. The definition of “nearby” (e.g., 30 bp) is provided as a command-line argument, and so is the imputed dosage threshold used to determine when the probability of a nearby germline variant being present is sufficiently high to warrant masking (e.g., 0.1 for a conservative mask).
This step is computationally light: <1 hour per chromosome for a large biobank, with minimal memory use because input data are streamed.
The 8_make_imp_masks.sh script directs genotype mask files to be written to the output/near_imputed_masks/ subdirectory of the analysis directory.
This script invokes the call_CNVs executable to perform haplotype-informed CNV calling. Probe intensity measurements are modeled using predicted genotype clusters, and data across haplotype neighbors are integrated in a Bayesian manner within a hidden Markov model (in which states correspond to copy numbers). DEL and DUP calls are obtained from a Viterbi decoding performed for each haplotype of each individual.
The extent to which information is incorporated from more-distant haplotype neighbors (trading off detection sensitivity for rarer versus less-rare CNVs) is modulated by a command-line IBD-weighting parameter T that optimizes for detecting CNVs that arose roughly T generations ago. To obtain high sensitivity across the spectrum of CNV allele ages, the 9_call_CNVs.sh script performs CNV-calling using a range of IBD-weighting parameter values. CNV calls made across the set of IBD-weighting parameters can then be synthesized in downstream analysis.
An additional command-line parameter sets the maximum extent to which a single probe measurement in a single individual is allowed to influence a CNV call (trading off the potential for a probe to be strongly informative of CNV presence against robustness to aberrant probe intensity measurements arising from technical artifacts). This parameter is less critical to analysis, and setting a single value (e.g., 3e-3) is likely to suffice.
This step is computationally moderate: on the order of 1 hour per chromosome for a large biobank (assuming multithreading is used), with higher memory use (up to 100 GB for a large biobank).
The 9_call_CNVs.sh script directs haplotype-informed CNV calls to be written to the output/HI-CNV/ subdirectory of the analysis directory.
The raw output produced by the final step of the above pipeline contains one line per CNV call with the following tab-delimited fields:
Note that the raw CNV call files described above are not yet suitable for downstream analyses! The raw calls are generated using very permissive thresholds, the idea being to supply a maximally comprehensive call set (likely containing many false positive calls) that can then be carefully post-processed.
Two main post-processing steps are necessary: First, further filtering needs to be performed to impose appropriate minimum thresholds on CNV call evidence (i.e., LOD = log10 Bayes factor) and on CNV length. Second, CNV calls need to be merged, both because (i) longer CNVs are sometimes fragmented into multiple shorter CNV calls separated by short gaps; and (ii) calls made for the same sample using different values of the IBD-weight parameter need to be synthesized.
Because the optimum parameter choices for filtering and merging may vary across data sets and may ideally be tuned using validation data (e.g., from whole-genome sequencing of a subset of samples), we have provided a separate set of post-processing scripts (in the post_processing subdirectory of the download package) that implement filtering and merging of raw calls.
Detailed descriptions of these R scripts together with example invocations are provided in the post_processing/README.txt file. Briefly, the scripts produce a few directories containing different sets of post-processed CNV calls, the most useful of which will likely be the deduped/ and flattened/ directories:
The output files in these directories contain one line per CNV call with the following tab-delimited fields:
All fields are described above, except “gt” which denotes the genotype (1 or 2) of each deduped call.
If you have comments or questions about the HI-CNV software, please contact:
Margaux Hujoel, mhujoel@broadinstitute.org
Po-Ru Loh, poruloh@broadinstitute.org
HI-CNV is free software under the GNU General Public License v3.0 (GPLv3).